Performance comparison of r2SCAN and SCAN metaGGA density functionals for solid materials via an automated, high-throughput computational workflow
Abstract
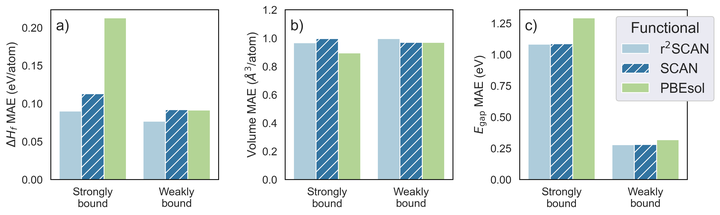
Computational materials discovery efforts utilize hundreds or thousands of density functional theory (DFT) calculations to predict material properties. Historically, such efforts have performed calculations at the generalized gradient approximation (GGA) level of theory due to its efficient compromise between accuracy and computational reliability. However, high-throughput calculations at the higher metaGGA level of theory are becoming feasible. The Strongly Constrainted and Appropriately Normed (SCAN) metaGGA functional offers superior accuracy to GGA across much of chemical space, making it appealing as a general-purpose metaGGA functional, but it suffers from numerical instabilities that impede it’s use in high-throughput workflows. The recently-developed r2SCAN metaGGA functional promises accuracy similar to SCAN in addition to more robust numerical performance. However, its performance compared to SCAN has yet to be evaluated over a large group of solid materials. In this work, we compared r2SCAN and SCAN predictions for key properties of approximately 6,000 solid materials using a newly-developed high-throughput computational workflow. We find that r2SCAN predicts formation energies more accurately than SCAN and PBEsol for both strongly- and weakly-bound materials and that r2SCAN predicts systematically larger lattice constants than SCAN. We also find that r2SCAN requires modestly fewer computational resources than SCAN and offers significantly more reliable convergence. Thus, our large-scale benchmark confirms that r2SCAN has delivered on its promises of numerical efficiency and accuracy, making it a preferred choice for high-throughput metaGGA calculations.